

La selección inadecuada de prensaestopas en entornos médicos y de salas limpias provoca riesgos de contaminación, fallos en el cumplimiento de la normativa y averías en los equipos que se traducen en riesgos para la seguridad de los pacientes, costosos cierres de las instalaciones e infracciones de la FDA, mientras que un sellado inadecuado, unos materiales inapropiados y una limpieza deficiente provocan la proliferación de bacterias, la contaminación por partículas y fallos de esterilidad que ponen en peligro las operaciones sanitarias críticas. Muchos gestores de instalaciones se esfuerzan por seleccionar prensaestopas que cumplan las estrictas normas médicas y, al mismo tiempo, mantengan un rendimiento eléctrico fiable.
La selección de prensaestopas para equipos médicos y salas blancas exige conocer la normativa de la FDA, USP Clase VI1 con construcción de acero inoxidable de grado médico o polímero especializado que proporciona biocompatibilidad, resistencia química y superficies lisas para una limpieza y esterilización eficaces, al tiempo que mantiene la integridad eléctrica en aplicaciones sanitarias críticas. El éxito depende de equilibrar el cumplimiento de la normativa con la fiabilidad operativa.
Tras haber trabajado con ingenieros de hospitales de los principales centros médicos de Boston, fabricantes farmacéuticos de Suiza e instalaciones de salas blancas de Singapur, he aprendido que los prensaestopas de calidad médica son esenciales para mantener entornos estériles y garantizar la seguridad de los pacientes. Permítame compartir con usted los conocimientos fundamentales para seleccionar los prensaestopas óptimos para sus aplicaciones médicas y de sala blanca.
Índice
- ¿Qué diferencia a los prensaestopas médicos de los prensaestopas estándar?
- ¿Cómo cumplir la normativa de la FDA y la de productos sanitarios?
- ¿Qué materiales se necesitan para las aplicaciones en salas blancas?
- ¿Cómo garantizar una correcta compatibilidad de limpieza y esterilización?
- ¿Cuáles son los principales criterios de selección para las distintas aplicaciones médicas?
- Preguntas frecuentes sobre prensaestopas médicos
¿Qué diferencia a los prensaestopas médicos de los prensaestopas estándar?
Los prensaestopas médicos se diferencian de los prensaestopas estándar en que utilizan materiales biocompatibles, presentan superficies lisas que se pueden limpiar, cumplen la normativa de la FDA, proporcionan un sellado mejorado para prevenir la contaminación y ofrecen resistencia química a los agentes de limpieza y los procesos de esterilización, con diseños especializados que eliminan las grietas donde podrían alojarse las bacterias, al tiempo que mantienen el rendimiento eléctrico en entornos sanitarios críticos.
Comprender estas diferencias es crucial porque las aplicaciones médicas tienen requisitos de seguridad y normativos únicos que los prensaestopas industriales estándar no pueden abordar adecuadamente.
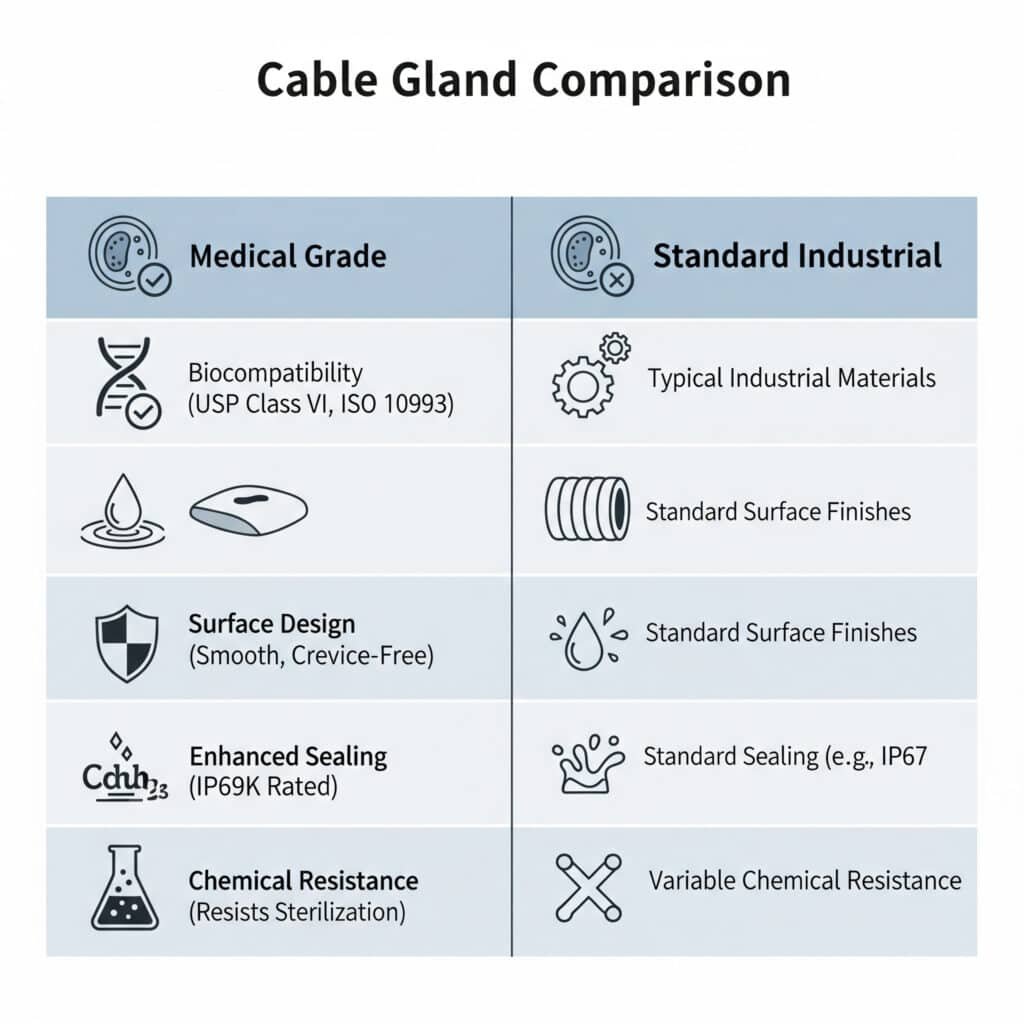
Requisitos de biocompatibilidad
Conformidad con USP Clase VI: Los prensaestopas médicos deben utilizar materiales que superen las pruebas biológicas de clase VI de la Farmacopea de los Estados Unidos para comprobar su biocompatibilidad y seguridad en aplicaciones de productos sanitarios.
Normas ISO 10993: Los materiales deben cumplir ISO 109932 las normas de evaluación biológica de los productos sanitarios, que garantizan la ausencia de efectos citotóxicos, sensibilizantes o irritantes en los tejidos humanos.
Aprobación FDA 21 CFR 177: Los materiales aptos para uso alimentario que cumplen la normativa de la FDA sobre contacto directo e indirecto con alimentos suelen ser necesarios para aplicaciones farmacéuticas y de dispositivos médicos.
Polímeros biocompatibles: Materiales especializados como el PEEK de grado médico, el PTFE o los nilones de grado farmacéutico proporcionan biocompatibilidad sin comprometer las propiedades mecánicas.
Superficies fáciles de limpiar
Superficie lisa Acabado: Los prensaestopas médicos presentan superficies ultrasuaves con valores Ra normalmente inferiores a 0,8 micrómetros para evitar la adhesión bacteriana y permitir una limpieza eficaz.
Diseño sin grietas: Eliminación de esquinas afiladas, roscas profundas y geometrías complejas que podrían albergar bacterias o resistir los procesos de limpieza y esterilización.
Bordes redondeados: Todas las superficies externas presentan bordes redondeados y transiciones suaves para facilitar la limpieza y evitar que se dañen los guantes de sala limpia o los envases estériles.
Superficie mínima: Los diseños aerodinámicos reducen al mínimo la superficie expuesta a la contaminación, al tiempo que mantienen las prestaciones mecánicas y eléctricas necesarias.
Mayor rendimiento de sellado
Clasificación IP68+: Protección medioambiental superior a la estándar IP68, alcanzando a menudo IP69K para aplicaciones de limpieza a alta presión y alta temperatura.
Barreras contra la contaminación: Las múltiples etapas de sellado evitan la entrada de partículas, bacterias y productos químicos de limpieza que podrían comprometer los entornos estériles.
Resistencia a la presión: Presiones nominales mejoradas para soportar procedimientos de limpieza y procesos de esterilización agresivos sin que se produzcan fallos de estanquidad.
Fiabilidad a largo plazo: Los materiales de sellado mantienen su integridad a través de repetidos ciclos de esterilización y exposición a productos químicos de limpieza agresivos.
Propiedades de resistencia química
Compatibilidad con agentes de limpieza: Resistencia a desinfectantes de uso hospitalario, compuestos de amonio cuaternario, peróxido de hidrógeno y otros productos químicos de limpieza agresivos.
Resistencia a la esterilización: Los materiales resisten la radiación gamma, el óxido de etileno, el autoclave de vapor y otros métodos de esterilización sin degradarse.
Productos químicos farmacéuticos: Resistencia a disolventes, ácidos, bases y compuestos farmacéuticos habituales en entornos de fabricación médica.
Estabilidad térmica: Mantener las propiedades mediante ciclos de temperatura de esterilización y requisitos de control de la temperatura de la sala blanca.
David, director de instalaciones de una importante fábrica farmacéutica de Nueva Jersey, se enfrentaba a problemas recurrentes de contaminación en sus líneas de llenado estéril, donde los prensaestopas estándar albergaban bacterias a pesar de los rigurosos protocolos de limpieza. Los prensaestopas de latón existentes tenían roscas y acabados superficiales complejos que no podían esterilizarse adecuadamente, lo que provocaba rechazos de lotes y problemas de conformidad con la FDA. Especificamos prensaestopas de acero inoxidable para uso médico con superficies electropulidas y diseños sin hendiduras que cumplían los requisitos USP Clase VI. La actualización eliminó las fuentes de contaminación, logró una eficacia de limpieza del 99,9% y ayudó a la instalación a superar las inspecciones de la FDA, al tiempo que redujo los índices de rechazo de productos en 85%. 😊
¿Cómo cumplir la normativa de la FDA y la de productos sanitarios?
Para cumplir las normativas de la FDA y de productos sanitarios es necesario utilizar materiales con las certificaciones adecuadas, mantener una documentación detallada, seguir Buenas prácticas de fabricación (BPF)3Los prensaestopas médicos requieren materiales aprobados por la FDA, pruebas de biocompatibilidad y controles de fabricación que garanticen una calidad y seguridad constantes en las aplicaciones de productos sanitarios.
El cumplimiento de la normativa no es negociable en las aplicaciones médicas porque los fallos pueden provocar daños a los pacientes, retiradas de productos y graves consecuencias legales.
Requisitos de material de la FDA
21 CFR Parte 177: Los materiales en contacto con productos farmacéuticos o dispositivos médicos deben cumplir la normativa sobre aditivos alimentarios de la FDA para sustancias en contacto indirecto con alimentos.
Fichero Maestro de Acceso (FMA): Los proveedores deben mantener archivos de acceso maestro de la FDA que documenten la seguridad de los materiales, los procesos de fabricación y los procedimientos de control de calidad.
Drug Master File (DMF): Para aplicaciones farmacéuticas, los materiales pueden requerir el registro en el Drug Master File con información detallada sobre composición y fabricación.
Certificado de conformidad: Los proveedores deben presentar certificados que confirmen que los materiales cumplen todas las normativas y especificaciones aplicables de la FDA.
Normas de calidad de los productos sanitarios
Cumplimiento de la norma ISO 13485: La fabricación debe seguir los sistemas de gestión de calidad de productos sanitarios ISO 13485 para el diseño, la producción y la vigilancia postcomercialización.
21 CFR Parte 820 (QSR): Cumplimiento de la normativa del sistema de calidad garantizando controles de diseño adecuados, control de documentos y acciones correctivas/preventivas.
Gestión de riesgos: Procesos de gestión de riesgos ISO 14971 para productos sanitarios, incluidos el análisis de riesgos, la evaluación y las medidas de control.
Controles de diseño: Procesos formales de control del diseño, incluida la planificación del diseño, los requisitos de entrada/salida, las revisiones, la verificación y la validación.

Documentación y trazabilidad
Certificados de materiales: Trazabilidad completa del material con certificados de análisis, resultados de pruebas de biocompatibilidad y documentación de cumplimiento normativo.
Registros de fabricación: Registros de fabricación detallados que incluyen parámetros de proceso, pruebas de control de calidad y documentación de lotes para una trazabilidad completa.
Control de cambios: Procedimientos formales de control de cambios para cualquier modificación de materiales, procesos o especificaciones que afecte al cumplimiento de la normativa.
Calificación de proveedores: Amplios programas de cualificación de proveedores que garantizan que todos los materiales y componentes cumplen los requisitos de los productos sanitarios.
Requisitos de validación
Pruebas de biocompatibilidad: Pruebas USP Clase VI que incluyen pruebas de inyección sistémica, intracutánea y de implantación para verificar la seguridad biológica.
Validación de la limpieza: Procedimientos de limpieza documentados con datos de validación que demuestren la eliminación eficaz de contaminantes y la reducción de la carga biológica.
Validación de la esterilización: Validación de los procesos de esterilización, incluidos el mapeo de dosis, los niveles de garantía de esterilidad y los estudios de compatibilidad de materiales.
Pruebas de rendimiento: Pruebas eléctricas, mecánicas y medioambientales para verificar que el rendimiento cumple los requisitos de los productos sanitarios durante todo el ciclo de vida del producto.
Apoyo a la presentación de solicitudes reglamentarias
Documentación 510(k): Documentación técnica de apoyo a las solicitudes 510(k) de la FDA para productos sanitarios que incorporan prensaestopas.
Expedientes técnicos: Expedientes técnicos completos para el marcado CE con arreglo al Reglamento sobre productos sanitarios (MDR) en los mercados europeos.
Acuerdos de calidad: Acuerdos formales de calidad con los proveedores en los que se definan responsabilidades, especificaciones y requisitos de cumplimiento.
Apoyo a la auditoría: Apoyo a las auditorías de la FDA, los organismos notificados y los clientes, incluida la revisión de la documentación y las inspecciones de las instalaciones.
¿Qué materiales se necesitan para las aplicaciones en salas blancas?
Las aplicaciones de salas limpias requieren materiales con propiedades de baja desgasificación, resistencia a la generación de partículas, compatibilidad química con los agentes de limpieza, superficies lisas no porosas y niveles de conductividad adecuados. El acero inoxidable 316L de calidad médica, el PEEK, el PTFE y los polímeros especializados aprobados para salas limpias proporcionan un rendimiento óptimo al tiempo que cumplen los requisitos. ISO 146444 normas de salas blancas y mantenimiento de la integridad eléctrica en entornos controlados.
La selección del material es fundamental porque las salas blancas requieren un control estricto de la contaminación por partículas y moléculas que podría comprometer la calidad del producto o las condiciones de esterilidad.
Requisitos del acero inoxidable
316L Grado Médico: Acero inoxidable austenítico con bajo contenido en carbono que proporciona una resistencia superior a la corrosión y biocompatibilidad para aplicaciones médicas.
Acabado electropulido: El electropulido elimina las imperfecciones de la superficie, reduce la generación de partículas y crea superficies lisas para una limpieza eficaz.
Tratamiento de pasivación: La pasivación química mejora la resistencia a la corrosión y elimina la contaminación por hierro que podría causar decoloración o generación de partículas.
Rugosidad superficial: Los valores Ra inferiores a 0,8 micrómetros (32 micropulgadas) minimizan la adherencia bacteriana y facilitan los procesos de limpieza y esterilización.
Polímeros de alto rendimiento
PEEK (Polieteretercetona): Su excelente resistencia química, baja desgasificación y biocompatibilidad hacen que el PEEK sea ideal para aplicaciones farmacéuticas y de dispositivos médicos.
PTFE (Politetrafluoroetileno): Su inercia química superior y sus propiedades antiadherentes proporcionan una excelente resistencia a los productos químicos de limpieza y a los materiales biológicos.
Nylon de grado médico: Los nilones especialmente formulados con homologación USP Clase VI ofrecen buenas propiedades mecánicas con biocompatibilidad para aplicaciones médicas.
POM aprobado para salas limpias: Polioximetileno con baja generación de partículas y buena estabilidad dimensional para aplicaciones de precisión en salas blancas.
Compatibilidad con la clasificación de salas limpias
ISO Clase 5 (Clase 100): Superficies ultrasuaves con mínima generación de partículas para aplicaciones de fabricación de semiconductores y productos farmacéuticos.
ISO Clase 6 (Clase 1000): Control moderado de partículas para la fabricación de dispositivos médicos y algunos procesos farmacéuticos.
ISO Clase 7 (Clase 10000): Requisitos estándar de las salas blancas para operaciones generales de montaje de productos farmacéuticos y sanitarios.
ISO Clase 8 (Clase 100000): Requisitos básicos de las salas blancas para el envasado y algunos procesos de fabricación de productos sanitarios.
Control de la desgasificación y la contaminación
Materiales de baja emisión de gases: Materiales con emisiones mínimas de compuestos orgánicos volátiles (COV) que puedan contaminar procesos o productos sensibles.
Contaminación molecular: Control de contaminantes moleculares como siliconas, plastificantes y otros compuestos orgánicos que podrían afectar a la calidad del producto.
Contaminación iónica: Materiales con bajo contenido iónico para evitar la contaminación de componentes electrónicos y productos farmacéuticos.
Sustancias extraíbles: Mínimas sustancias extraíbles que puedan filtrarse en productos farmacéuticos o soluciones de limpieza durante su uso.
Matriz de compatibilidad química
| Agente de limpieza | ACERO INOXIDABLE 316L | PEEK | PTFE | Nylon médico |
|---|---|---|---|---|
| Alcohol isopropílico | Excelente | Excelente | Excelente | Bien |
| Peróxido de hidrógeno | Excelente | Excelente | Excelente | Feria |
| Amonio cuaternario | Excelente | Excelente | Excelente | Bien |
| Hipoclorito sódico | Bien | Excelente | Excelente | Pobre |
| Ácido peracético | Bien | Excelente | Excelente | Pobre |
Hassan, que gestiona las operaciones de unas instalaciones farmacéuticas de vanguardia en Suiza, necesitaba actualizar los prensaestopas de su sala blanca ISO Clase 5 para la fabricación de inyectables estériles. Los prensaestopas existentes generaban partículas durante los ciclos de limpieza y no soportaban los agresivos protocolos de esterilización necesarios para sus nuevos productos biológicos. Suministramos prensaestopas de acero inoxidable 316L electropulido con baja generación de partículas validada y total compatibilidad química con sus procesos de limpieza y esterilización. La mejora consiguió un recuento de partículas 90% por debajo de los límites de la norma ISO Clase 5, eliminó los casos de contaminación relacionados con la limpieza y contribuyó al éxito de la validación de su nueva línea de fabricación estéril.
¿Cómo garantizar una correcta compatibilidad de limpieza y esterilización?
Para garantizar una compatibilidad adecuada de la limpieza y la esterilización es necesario seleccionar materiales que resistan la exposición repetida a los productos químicos de limpieza y a los métodos de esterilización, diseñar superficies que se puedan limpiar completamente, validar los procedimientos de limpieza y mantener una documentación detallada. Los prensaestopas médicos requieren superficies lisas y sin grietas, materiales resistentes a los productos químicos y protocolos de limpieza validados que alcancen los niveles de garantía de esterilidad requeridos.
La compatibilidad de la limpieza y la esterilización es esencial porque una descontaminación inadecuada puede provocar la contaminación del producto, riesgos para la seguridad del paciente e infracciones de la normativa.
Compatibilidad del método de esterilización
Autoclave de vapor: Los materiales deben resistir 121°C-134°C autoclave con vapor5 ciclos sin degradación, cambios dimensionales o fallo de la junta.
Radiación gamma: Resistencia a dosis de radiación gamma de 25-50 kGy utilizadas habitualmente para la esterilización de dispositivos médicos sin degradación del material.
Óxido de etileno (EtO): Compatibilidad química con la esterilización por EtO, incluida la resistencia al esterilizante y las características adecuadas de desgasificación.
Plasma de peróxido de hidrógeno: Compatibilidad con los sistemas de esterilización por plasma a baja temperatura, incluida la estabilidad del material y la penetración completa del esterilizante.
Requisitos de validación de la limpieza
Procedimientos de limpieza: Procedimientos de limpieza documentados que especifiquen los productos químicos, las concentraciones, los tiempos de contacto y la acción mecánica necesarios para una descontaminación eficaz.
Protocolos de validación: Estudios de validación formal que demuestren la eficacia de la limpieza utilizando los peores escenarios de contaminación y pruebas analíticas.
Criterios de aceptación: Criterios de aceptación definidos para la limpieza, incluida la inspección visual, el recuento de partículas, los niveles de carga biológica y los límites de residuos químicos.
Control rutinario: Programas de supervisión continua para verificar la eficacia de la limpieza e identificar cualquier degradación del rendimiento.
Consideraciones sobre el diseño de superficies
Características del drenaje: Características de diseño que favorecen el drenaje completo de las soluciones de limpieza y evitan la formación de charcos que podrían albergar contaminantes.
Accesibilidad: Todas las superficies deben ser accesibles para su limpieza con las herramientas y procedimientos de limpieza estándar utilizados en las instalaciones médicas.
Requisitos de desmontaje: Considere si es necesario desmontar los prensaestopas para su limpieza o si la limpieza intacta es suficiente para la aplicación.
Sustitución de juntas: Procedimientos para la sustitución y validación de juntas cuando los componentes de sellado requieren una sustitución periódica debido a la exposición a productos químicos de limpieza.
Documentación y conformidad
Instrucciones de limpieza: Instrucciones detalladas de limpieza que incluyen procedimientos paso a paso, especificaciones químicas y precauciones de seguridad.
Datos de seguridad: Información completa sobre la seguridad de los materiales, incluida la compatibilidad química, los límites de temperatura y la compatibilidad con la esterilización.
Informes de validación: Informes de validación de la limpieza que demuestren la eficacia y establezcan los requisitos de seguimiento rutinario.
Material de formación: Material de formación para el personal de las instalaciones sobre los procedimientos adecuados de limpieza, manipulación y mantenimiento de los prensaestopas médicos.
Pruebas de control de calidad
Pruebas de carga biológica: Pruebas periódicas de contaminación microbiana para verificar la eficacia de la limpieza e identificar posibles zonas problemáticas.
Pruebas de endotoxinas: Pruebas de endotoxinas bacterianas que podrían causar reacciones pirogénicas en aplicaciones farmacéuticas y de dispositivos médicos.
Recuento de partículas: Pruebas de recuento de partículas para verificar que se mantienen los requisitos de la sala limpia tras las actividades de limpieza y mantenimiento.
Análisis de residuos químicos: Pruebas de residuos químicos de limpieza que podrían contaminar los productos o afectar a la biocompatibilidad.
¿Cuáles son los principales criterios de selección para las distintas aplicaciones médicas?
Los criterios clave de selección varían en función de la aplicación, pero incluyen requisitos de cumplimiento de normativas, niveles de biocompatibilidad, clasificaciones de salas limpias, métodos de esterilización, exposición química y necesidades de rendimiento eléctrico; los equipos quirúrgicos requieren la máxima biocompatibilidad, la fabricación de productos farmacéuticos necesita resistencia química y los equipos de diagnóstico se centran en la integridad eléctrica, mientras que todas las aplicaciones requieren certificaciones normativas adecuadas y compatibilidad de limpieza.
Las distintas aplicaciones médicas tienen requisitos únicos que deben tenerse muy en cuenta durante la selección de los prensaestopas para garantizar un rendimiento óptimo y el cumplimiento de la normativa.
Equipos quirúrgicos y de contacto con el paciente
Requisitos de contacto directo: Biocompatibilidad USP Clase VI con pruebas adicionales de citotoxicidad para materiales en contacto directo con el paciente durante procedimientos quirúrgicos.
Frecuencia de esterilización: Capacidad para soportar frecuentes ciclos de esterilización sin degradación, lo que suele requerir materiales estables durante cientos de ciclos de esterilización.
Seguridad eléctrica: Requisitos de seguridad eléctrica mejorados, incluida una baja corriente de fuga y un aislamiento fiable para los equipos conectados a los pacientes.
Fiabilidad en caso de emergencia: Requisitos críticos de fiabilidad para equipos de soporte vital y emergencias médicas en los que un fallo podría afectar directamente a la seguridad del paciente.
Fabricación farmacéutica
Cumplimiento de las GMP: Requisitos de buenas prácticas de fabricación, incluida la trazabilidad de los materiales, el control de cambios y la documentación de validación.
Seguridad de contacto del producto: Materiales que no contaminen los productos farmacéuticos por lixiviación, generación de partículas o interacción química.
Validación de la limpieza: Amplios requisitos de validación de la limpieza con procedimientos documentados y criterios de aceptación para la fabricación de productos farmacéuticos.
Documentación de lotes: Documentación completa de los lotes y trazabilidad de los materiales utilizados en los equipos de fabricación farmacéutica.
Equipos de diagnóstico y laboratorio
Requisitos de precisión: Estabilidad dimensional y precisión para instrumentos analíticos que requieren un rendimiento mecánico y eléctrico constante.
Resistencia química: Resistencia a los productos químicos de laboratorio, reactivos y disolventes de limpieza utilizados habitualmente en aplicaciones de diagnóstico.
Rendimiento de EMC: Requisitos de compatibilidad electromagnética para instrumentos analíticos y equipos de diagnóstico sensibles.
Estabilidad de la calibración: Estabilidad mecánica que no afectará a la calibración de los instrumentos ni a la precisión de las mediciones con el paso del tiempo.
Fabricación de productos sanitarios
Validación del proceso: Materiales y componentes que cumplen los requisitos de validación de procesos para la fabricación de productos sanitarios.
Sistemas de calidad: Integración con los sistemas de calidad ISO 13485, incluida la cualificación de proveedores y los procedimientos de inspección de entrada.
Gestión de riesgos: Materiales de apoyo a los requisitos de gestión de riesgos de los productos sanitarios, incluido el análisis de modos de fallo y las estrategias de mitigación.
Apoyo normativo: Apoyo a los proveedores para la presentación de documentos reglamentarios, incluida la documentación técnica y los conocimientos reglamentarios.
Matriz de decisión para la selección
| Tipo de aplicación | Criterios primarios | Criterios secundarios | Normas críticas |
|---|---|---|---|
| Equipo quirúrgico | Biocompatibilidad | Esterilización | USP Clase VI, ISO 10993 |
| Farmacéutica | Resistencia química | Cumplimiento de las GMP | FDA 21 CFR, cGMP |
| Diagnóstico | Precisión | Rendimiento del EMC | IEC 61326, ISO 15189 |
| Fabricación | Validación del proceso | Sistemas de calidad | ISO 13485, 21 CFR 820 |
Análisis coste-beneficio
Coste inicial frente a ciclo de vida: Considere el coste total de propiedad, incluidos los costes de mantenimiento, sustitución y cumplimiento durante la vida útil del equipo.
Riesgo reglamentario: Equilibrar los costes materiales con los riesgos de cumplimiento de la normativa y los costes potenciales del incumplimiento.
Requisitos de rendimiento: Garantizar que los materiales seleccionados cumplen los requisitos mínimos de rendimiento sin un exceso de especificaciones que aumente los costes innecesariamente.
Capacidades de los proveedores: Evaluar las capacidades de los proveedores en materia de asistencia continua, documentación y cumplimiento de la normativa.
Conclusión
La selección de prensaestopas para equipos médicos y salas blancas exige conocer los requisitos normativos, de materiales y de rendimiento específicos que distinguen estas aplicaciones de los usos industriales estándar. El éxito depende del equilibrio entre biocompatibilidad, facilidad de limpieza, cumplimiento de la normativa y rendimiento eléctrico.
La clave para un rendimiento fiable de los prensaestopas médicos reside en trabajar con proveedores que conozcan la normativa sobre dispositivos médicos y puedan proporcionar los materiales adecuados, la documentación y la asistencia continua durante todo el ciclo de vida del producto. En Bepto, estamos especializados en prensaestopas de grado médico que cumplen los requisitos de la FDA y las normas internacionales sobre dispositivos médicos, proporcionando la garantía de calidad y el soporte normativo necesarios para aplicaciones sanitarias críticas.
Preguntas frecuentes sobre prensaestopas médicos
P: ¿Qué certificaciones necesito para los prensaestopas médicos?
A: Los prensaestopas médicos suelen requerir la certificación de biocompatibilidad USP Clase VI, la conformidad de los materiales con FDA 21 CFR y sistemas de calidad de fabricación ISO 13485. Los requisitos específicos dependen de su aplicación y de la jurisdicción reguladora.
P: ¿Pueden utilizarse los prensaestopas de acero inoxidable normales en aplicaciones médicas?
A: El acero inoxidable normal puede no cumplir los requisitos médicos. Las aplicaciones médicas requieren acero inoxidable 316L de calidad médica con acabado electropulido, pruebas de biocompatibilidad y la documentación adecuada para el cumplimiento de la normativa.
P: ¿Cómo se limpian y esterilizan los prensaestopas médicos?
A: Siga los procedimientos de limpieza validados por el fabricante utilizando agentes de limpieza y métodos de esterilización aprobados. La mayoría de las glándulas médicas admiten autoclave con vapor, radiación gamma o esterilización química con la compatibilidad de materiales adecuada.
P: ¿Qué diferencia hay entre los prensaestopas para sala blanca y los de uso médico?
A: El grado médico incluye requisitos de biocompatibilidad y conformidad con la FDA, mientras que el grado de sala limpia se centra en el control de partículas y la resistencia química. Algunas aplicaciones requieren tanto especificaciones médicas como de sala blanca.
P: ¿Con qué frecuencia deben sustituirse los prensaestopas médicos?
A: La frecuencia de sustitución depende de los ciclos de esterilización, la exposición química y las recomendaciones del fabricante. Normalmente se inspecciona cada 100-200 ciclos de esterilización y se sustituye en función de la evaluación del estado y los requisitos de validación.
-
Conozca las pruebas específicas de reactividad biológica requeridas para la certificación USP Clase VI. ↩
-
Acceda a una visión general de la serie de normas ISO 10993 para la evaluación biológica de productos sanitarios. ↩
-
Revise las directrices y reglamentos oficiales de la FDA para las Buenas Prácticas de Fabricación Actuales (cGMP). ↩
-
Explore una guía detallada de las normas ISO 14644 para clasificar la limpieza del aire en salas blancas. ↩
-
Comprender los principios de la esterilización por vapor, incluido el papel del tiempo, la temperatura y la presión. ↩
